Research
Genome-wide association studies (GWAS) have established a major role of non-coding variants in Age-related macular degeneration (AMD), which is the leading cause of blindness in the elderly. However, the biological interpretation of AMD-GWAS findings remains challenging as we have a limited understanding of how associated non-coding variants translate into disease pathogenesis. In recent years, gene expression regulation has emerged as a dominant mechanism for non-coding variants to mediate the disease risk. I aided functional interpretation of AMD-associated, non-coding variants by performing a large scale, integrative analyses of transcriptome (RNAseq) and their genetic variations from a large cohort (453 individuals) including both controls and cases at distinct stages of AMD. As next step, we generate and integrate multiple high-throughput functional genomics data sets on the genome, transcriptome, and epigenome, in disease-relevant tissues/cells and use computational approaches identify causal variant, disease-associated cis-regulatory regions, target gene, and the disease mechanism underlying a GWAS loci.
1. Ratnapriya R, Sosina OA, Starostik MR, Kwicklis M, Kapphahn RJ, Fritsche LG, Walton A, Arvanitis M, Gieser L, Pietraszkiewicz A, Montezuma SR, Chew EY, Battle A, Abecasis GR, Ferrington DA, Chatterjee N, Swaroop A. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet. 2019 Apr;51(4):606-610. doi: 10.1038/s41588-019-0351-9. Epub 2019 Feb 11. Erratum in: Nat Genet. 2019 Jun;51(6):1067. doi: 10.1038/s41588-019-0430-y. PMID: 30742112; PMCID: PMC6441365.
2. Ratnapriya R, Grassman F, Chen R, Hewitt A, Du J, Saban DR, Klaver CCW, Ash J, Stambolian D, Tumminia SJ, Qian J, Husain D, Iyengar SK, den Hollander AI. Functional genomics in age-related macular degeneration: From genetic associations to understanding disease mechanisms. Exp Eye Res. 2025 May;254:110344. doi: 10.1016/j.exer.2025.110344. Epub 2025 Mar 13. PMID: 40089136; PMCID: PMC12048874.
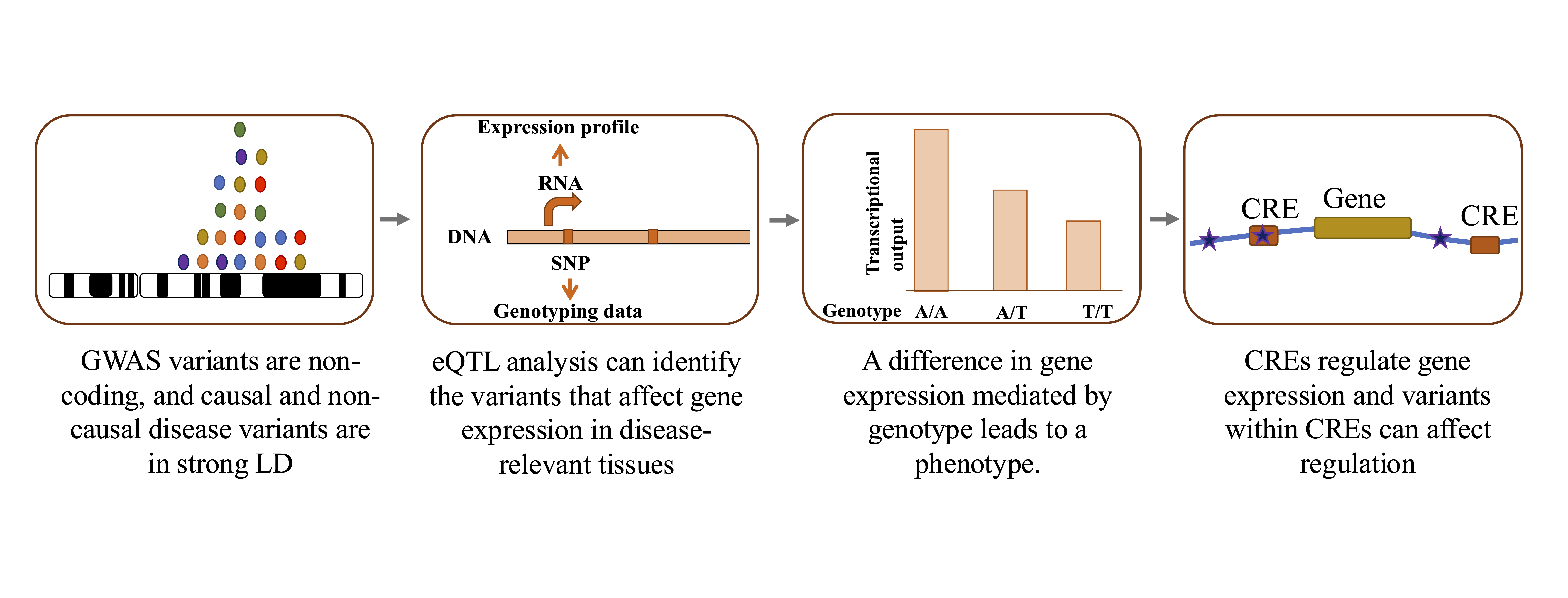
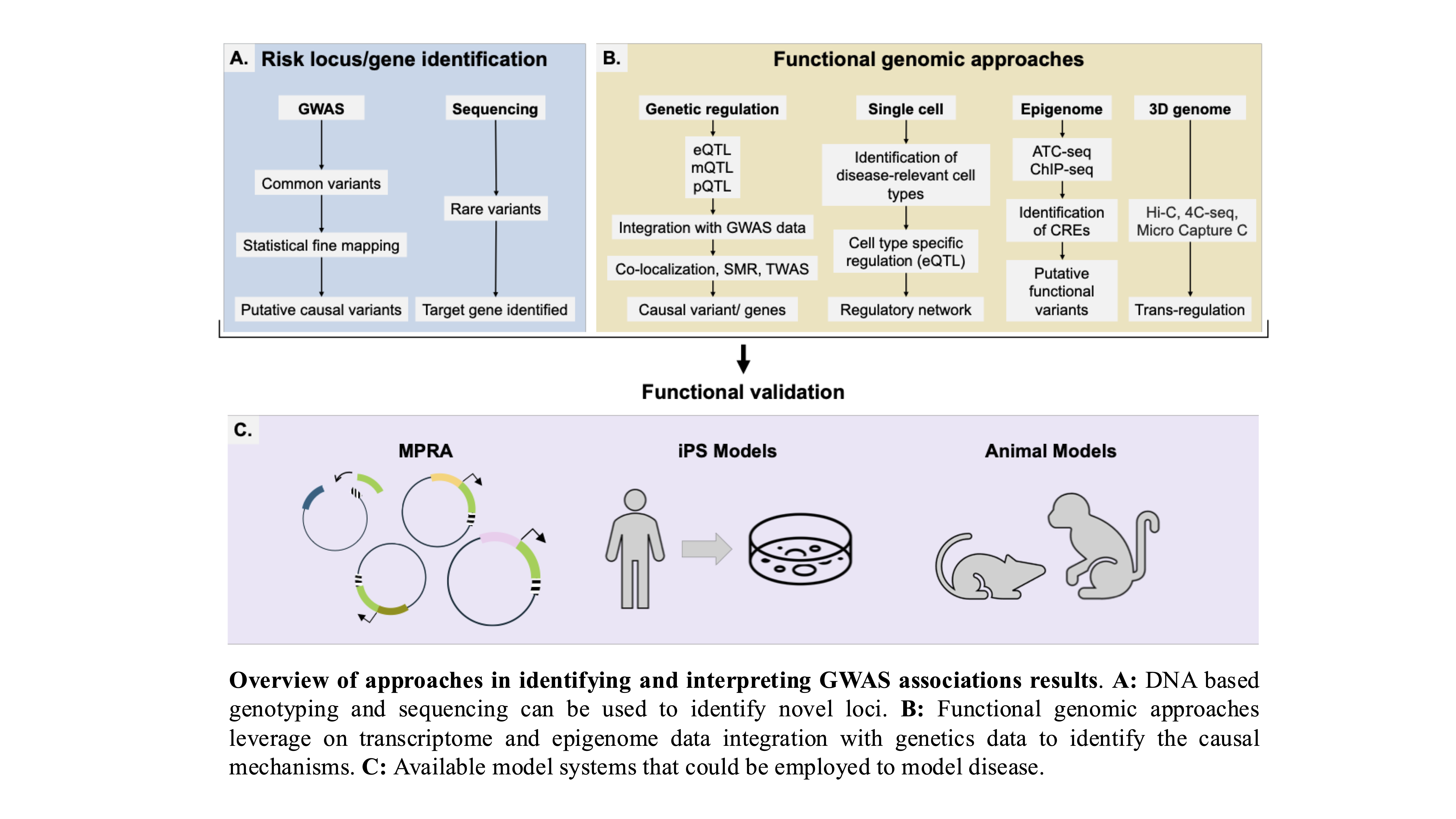
Variation in gene expression is a key driver of phenotypic diversity and plays a central role in the development of both rare and common diseases. Understanding gene expression dysregulation is crucial for uncovering molecular disease mechanisms and identifying therapeutic targets. However, studying gene expression in disease-relevant human tissues is challenging due to limited sample availability and the influence of genetic, epigenetic, and environmental factors.To address these challenges, we apply machine learning (ML) methods to uncover biologically meaningful patterns in transcriptomic data.
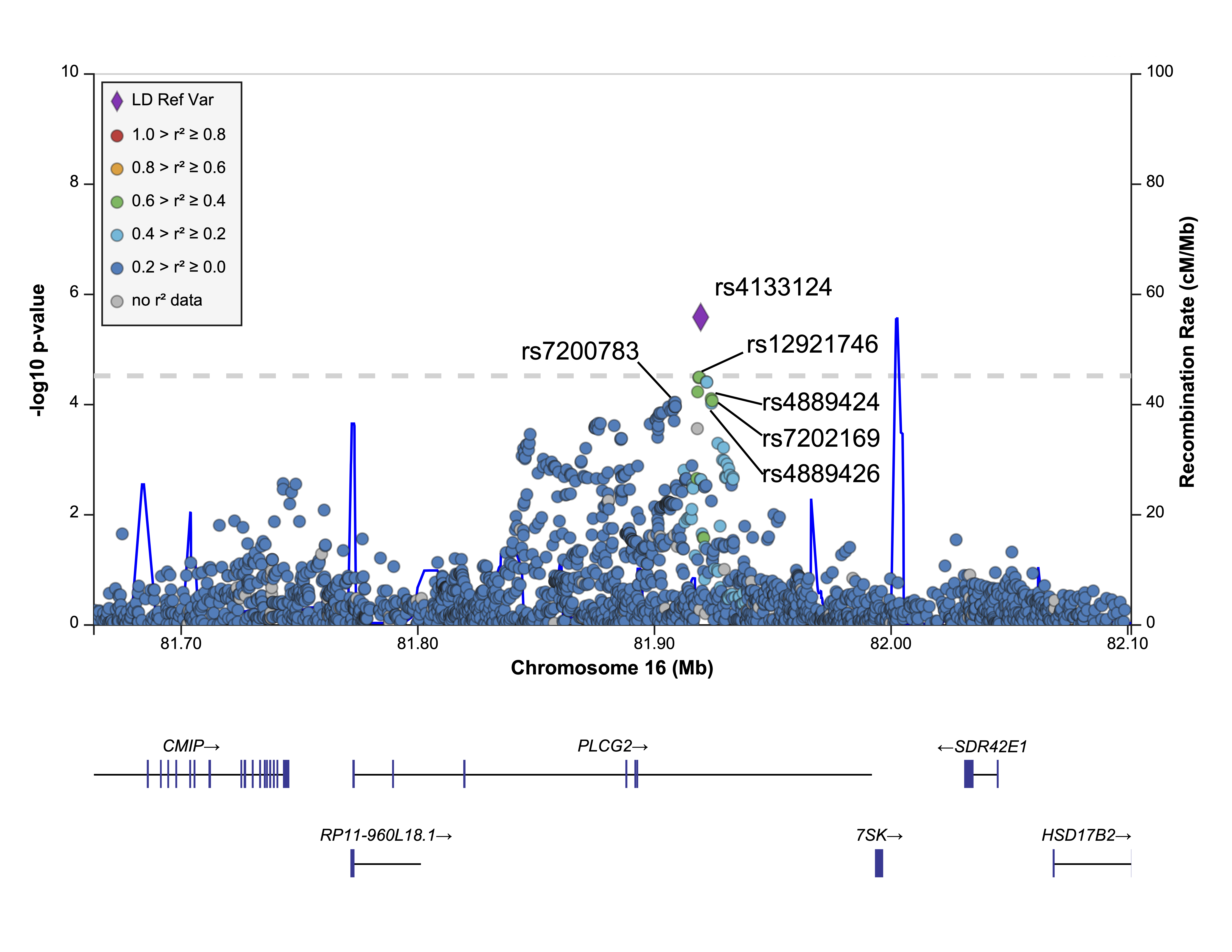
In a recent work, we have developed an explainable ML pipeline using retinal transcriptomes from 453 donors, identifying 81 genes that distinguish AMD patients from controls (AUC-ROC 0.80, CI 0.70–0.92). Our model leverages non-linear data relationships and employs rigorous feature selection and dimensionality reduction. Integrating AMD GWAS data, we also identified rs4133124 at PLCG2 as a novel regulatory variant associated with AMD. These results highlight the benefits of integrative analytical approaches to regain the holistic view of the AMD that is lost in experimentally tested reductionist approaches or hard statistical cut-offs.


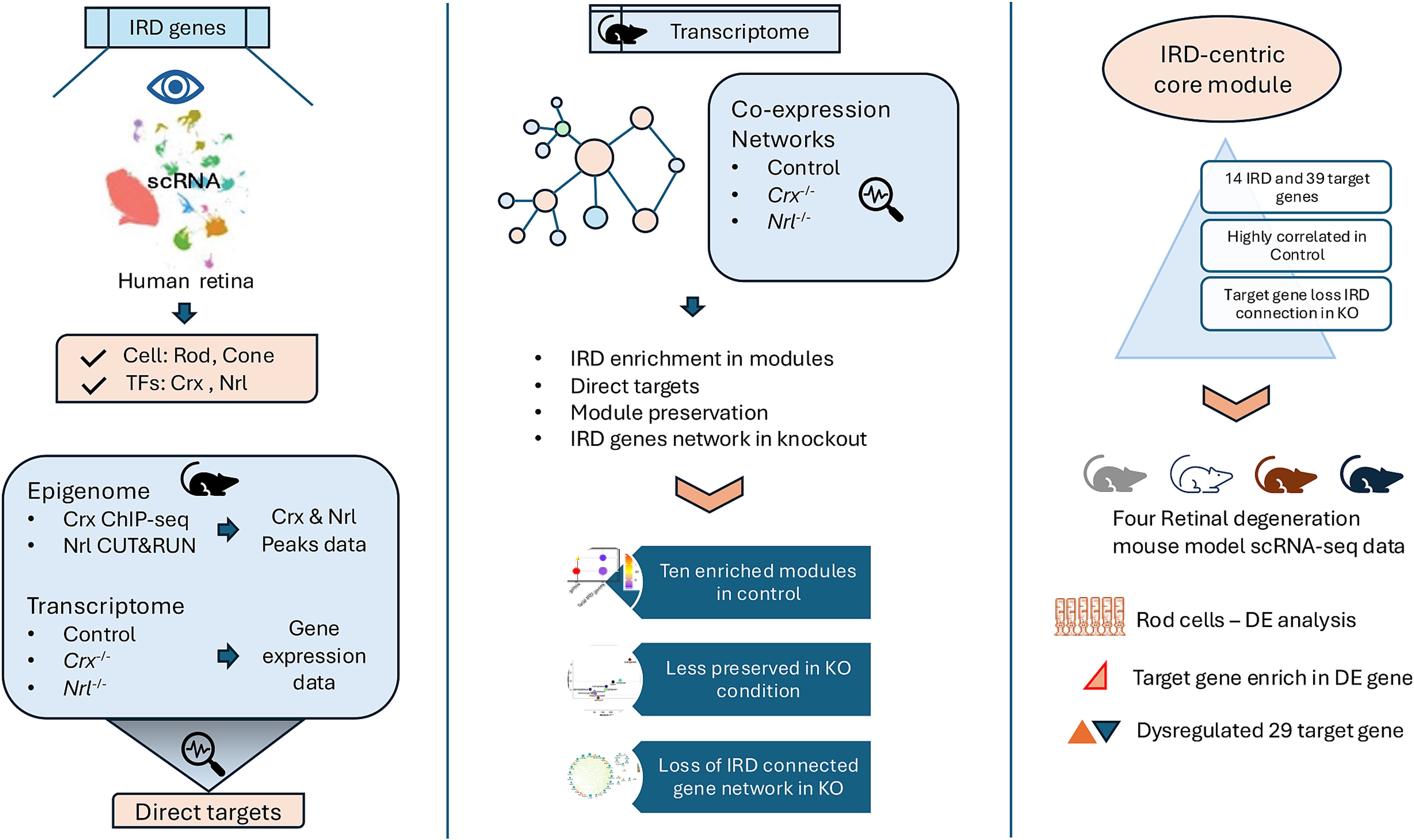
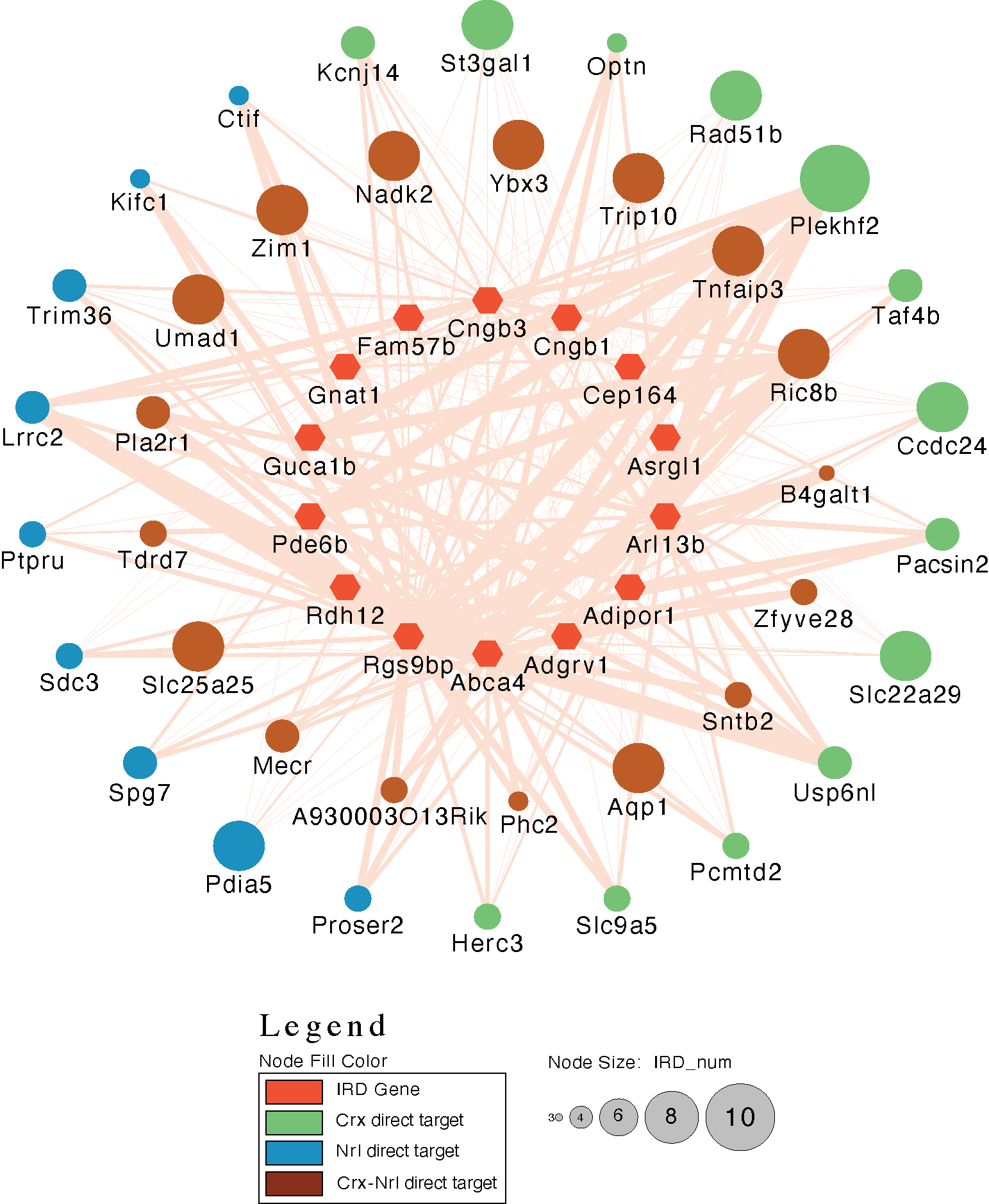
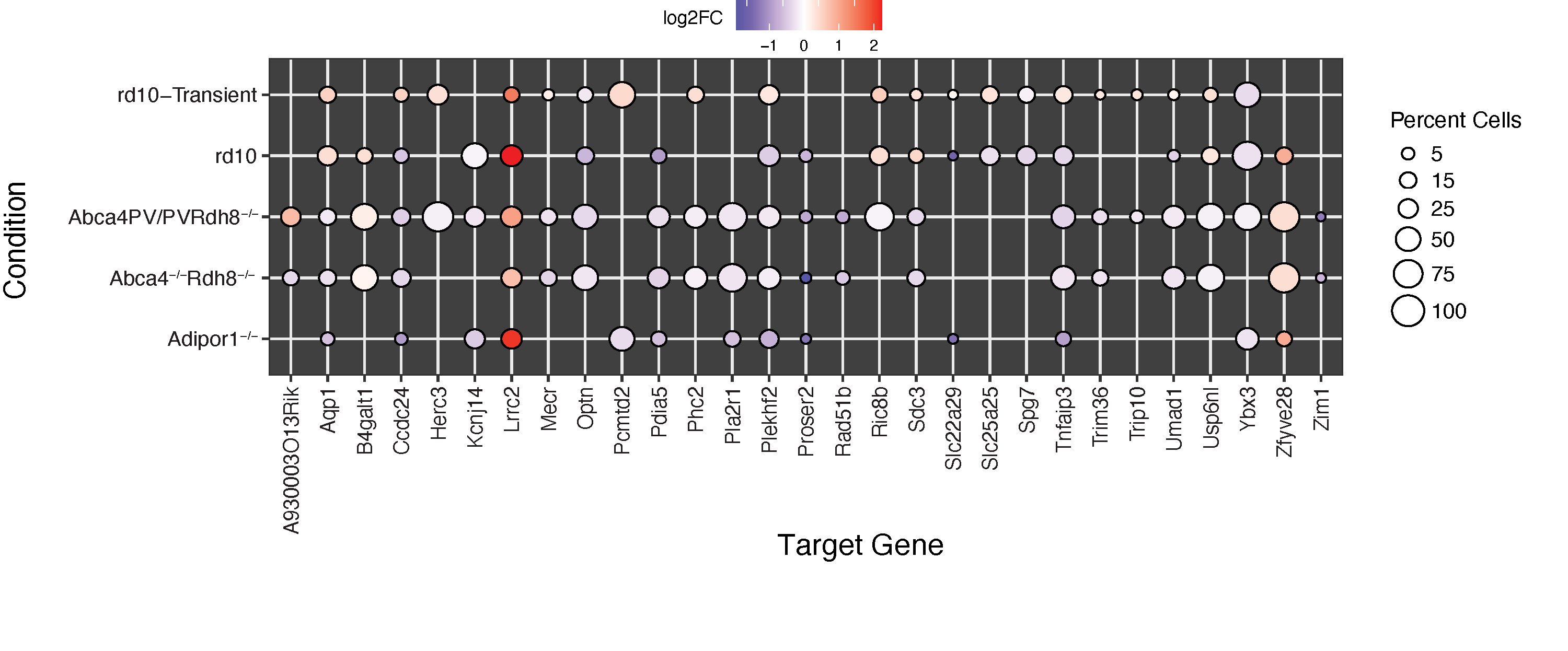
Human diseases with similar phenotypes can be interconnected through shared biological pathways, genes, or molecular mechanisms. Inherited retinal diseases (IRDs) cause photoreceptor dysfunction due to mutations in approximately 300 genes, affecting visual transduction, photoreceptor morphogenesis, and transcription factors, suggesting common pathobiological mechanisms. The high clinical heterogeneity of IRDs makes it challenging to develop effective therapeutic interventions in large cohorts of patients. Therefore, our lab is interested in applying genomic approaches to understand the functional relationships among IRD genes that culminate in photoreceptor degeneration.
In a recent study we examined the functional relationship between known IRDs genes by integrating peaks and gene expression data from the key photoreceptor transcription factors (TFs) Crx and Nrl. We demonstrated that the targets of these TFs were enriched in IRDs causal genes. Co-expression network analysis revealed that IRD-centric networks were disrupted when Crx and Nrl were knocked out. Finally, we identified a highly connected core module comprising 14 IRD and 39 target genes, of which 29 were dysregulated in the rod photoreceptors of the four IRD mouse models. These findings offer a network-based interpretation of IRDs, aiding in the identification of common mechanisms, prioritizing genes for novel disease gene identification, and informing the development of gene-agnostic therapies for IRDs.
Singh A, Ratnapriya R. Integration of multiomic data identifies core-module of inherited-retinal diseases. Hum Mol Genet. 2025 Feb 17;34(5):454-465. doi: 10.1093/hmg/ddaf001. PMID: 39797395
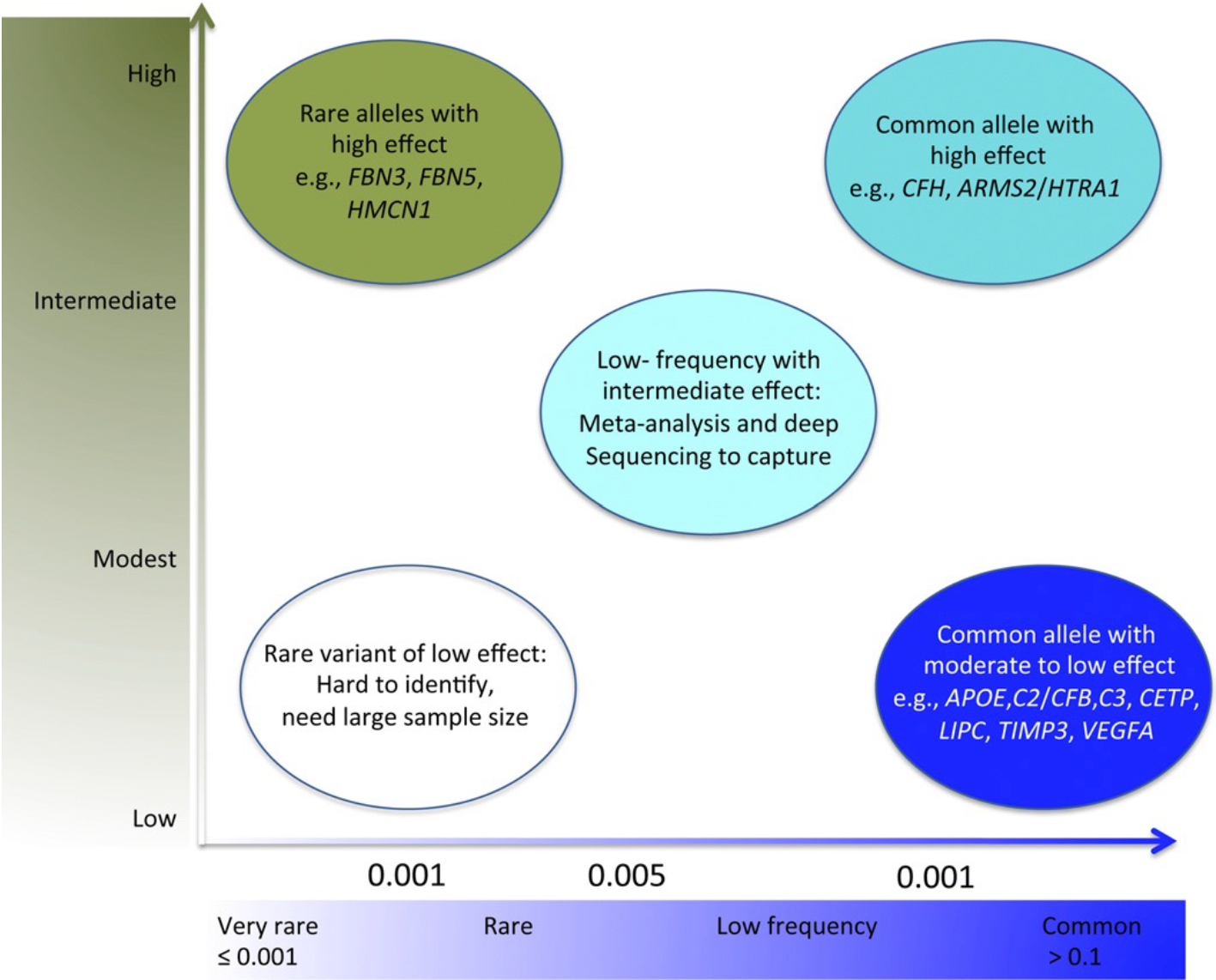
GWAS signals are primarily, common, non-coding variants and identity of the causal gene is not obvious . In a shift from common-disease common-variants (CDCV) hypothesis, the role of rare variants are being recognized in complex diseases. Identification of a rare variants can provide hypothesis-free evidence for causality by pointing to the target gene underlying AMD. Thus, we apply whole exome sequencing to identify the rare variants in AMD and related maculopathies. Our most recent work on the analysis of 63 multiplex AMD families identified rare variants and target genes at eight AMD loci.
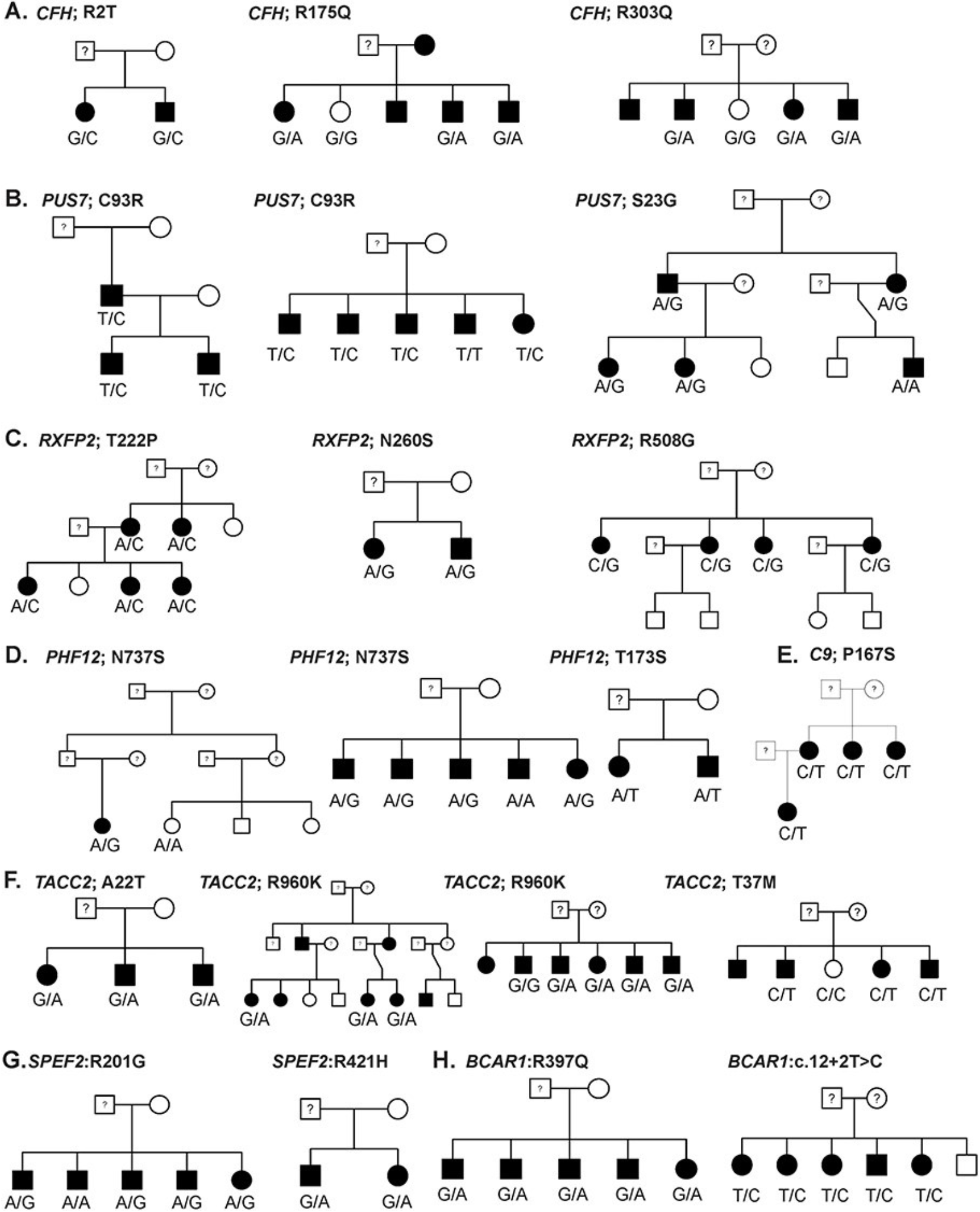
Mutations in >300 genes are associated with Inherited Retinal Degenerative disease (IRDs) and display considerable clinical heterogeneity. We apply whole-exome sequencing for rapid identification of disease-causing variants in retinal disease patients and families with the aim of expediting the novel disease gene discovery, improved molecular diagnosis, and for the understanding of the role of more than one gene in causing monogenic diseases.
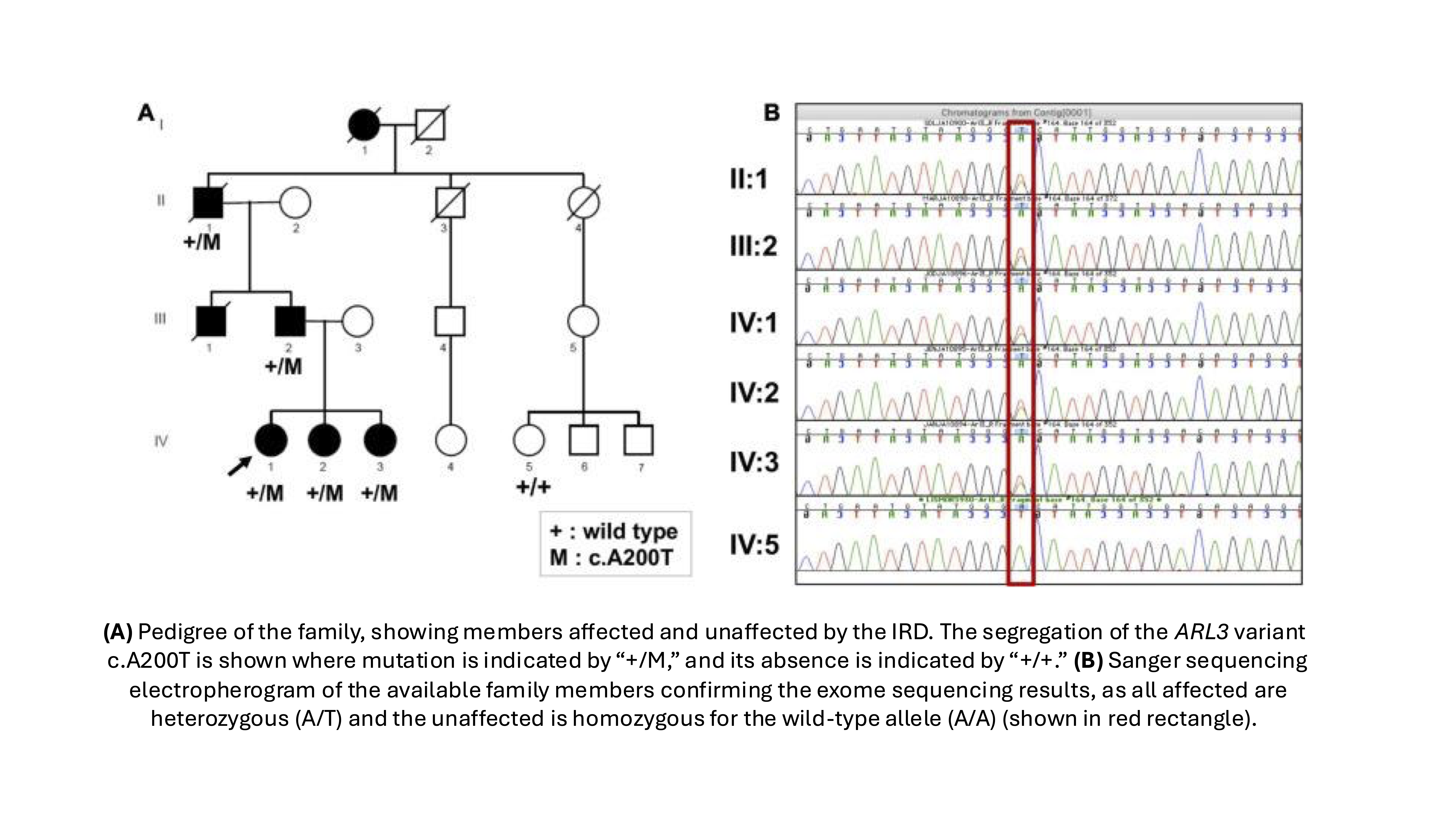
1. Ratnapriya R, Jacobson SG, Cideciyan AV, English MA, Roman AJ, Sumaroka A, Sheplock R, Swaroop A. A Novel ARL3 Gene Mutation Associated With Autosomal Dominant Retinal Degeneration. Front Cell Dev Biol. 2021 Aug 17;9:720782. doi: 10.3389/fcell.2021.720782. PMID: 34485303; PMCID: PMC8416110.
2. Pierrache LHM, Kimchi A, Ratnapriya R, Roberts L, Astuti GDN, Obolensky A, Beryozkin A, Tjon-Fo-Sang MJH, Schuil J, Klaver CCW, Bongers EMHF, Haer-Wigman L, Schalij N, Breuning MH, Fischer GM, Banin E, Ramesar RS, Swaroop A, van den Born LI, Sharon D, Cremers FPM. Whole-Exome Sequencing Identifies Biallelic IDH3A Variants as a Cause of Retinitis Pigmentosa Accompanied by Pseudocoloboma. Ophthalmology. 2017 Jul;124(7):992-1003. doi: 10.1016/j.ophtha.2017.03.010. Epub 2017 Apr 13. PMID: 28412069; PMCID: PMC5868413.
3. Roberts L, Ratnapriya R, du Plessis M, Chaitankar V, Ramesar RS, Swaroop A. Molecular Diagnosis of Inherited Retinal Diseases in Indigenous African Populations by Whole-Exome Sequencing. Invest Ophthalmol Vis Sci. 2016 Nov 1;57(14):6374-6381. doi: 10.1167/iovs.16-19785. PMID: 27898983; PMCID: PMC5132076.
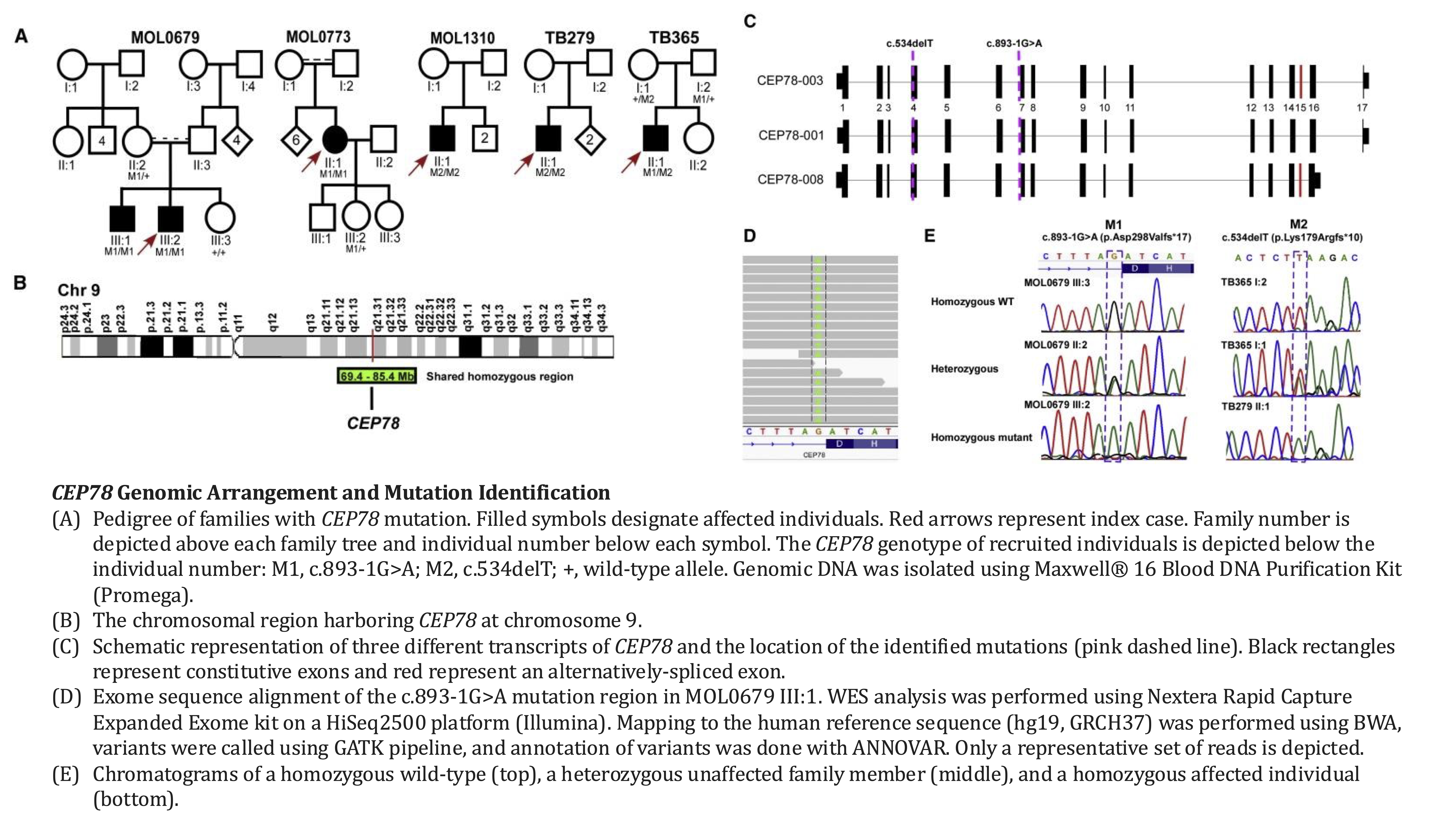
4. Namburi P, Ratnapriya R, Khateb S, Lazar CH, Kinarty Y, Obolensky A, Erdinest I, Marks-Ohana D, Pras E, Ben-Yosef T, Newman H, Gross M, Swaroop A, Banin E, Sharon D. Bi-allelic Truncating Mutations in CEP78, Encoding Centrosomal Protein 78, Cause Cone-Rod Degeneration with Sensorineural Hearing Loss. Am J Hum Genet. 2016 Nov 3;99(5):1222-1223. doi: 10.1016/j.ajhg.2016.09.012. Erratum for: Am J Hum Genet. 2016 Sep 1;99(3):777-784. doi: 10.1016/j.ajhg.2016.07.010. PMID: 27814526; PMCID: PMC5097977.
Contact
Social
© 2025 RatnapriyaLab. All rights reserved.